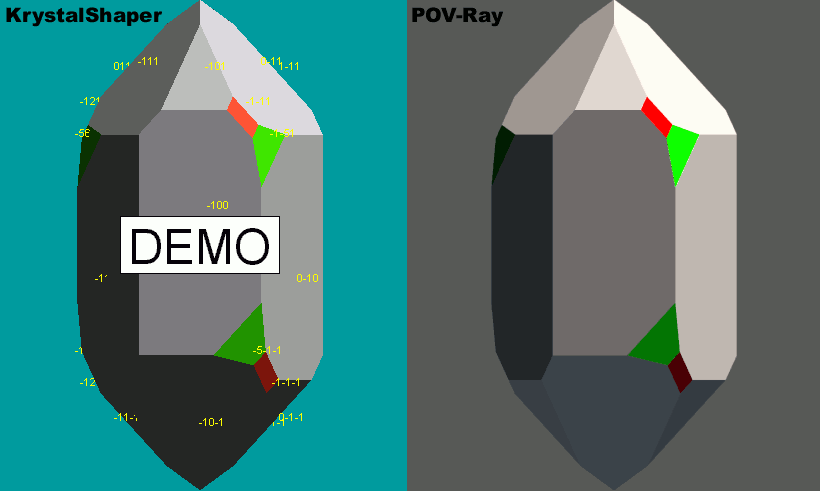
|
|
|
|
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Hi everyone!
No amazing eye candy to show you, but I have resolved the issues
surrounding my inability to accurately depict odd angles for certain
crystal morphologies.
Attached is a png image showing both a screenshot from KrystalShaper,
and a corresponding crystal shape rendered with POV-Ray. The problem
areas are highlighted in red and green.
I used exact angles and distances for this test. My reproduction seems
100% accurate, which means I can go on to create a library of minerals
with support for randomized distances. A tutorial describing the
technique may also be possible (although I still have that Weathered
Surfaces tutorial to finish).
Questions or comments are always welcome!
Sam
Post a reply to this message
Attachments:
Download 'quartz.png' (10 KB)
Preview of image 'quartz.png'
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Cool, I saw your hexagon and was messing with
Miller indices, and I found this site
http://www.gly.uga.edu/schroeder/geol6550/millerindices.html
which seems to explain simple Miller indices OK.
Still haven't looked at Miller-Bravais indices.
This is what I ended up with, but it's mostly untested.
#macro ThreePointPlane(P1, P2, P3)
#local n = vnormalize(vcross(P1-P2,P3-P2));
#local k = vdot(n,P1);
plane {n,k}
#end
#macro MillerPlane(l,m,n)
#if (l!=0) #local lp = <1/l,0,0>; #else #if (m!=0) #local lp = <1,0,1/m>;
#else #local lp = <1,1/n,0>; #end #end
#if (m!=0) #local mp = <0,0,1/m>; #else #if (l!=0) #local mp = <1/l,0,1>;
#else #local mp = <0,1/n,1>; #end #end
#if (n!=0) #local np = <0,1/n,0>; #else #if (l!=0) #local np = <1/l,1,0>;
#else #local np = <0,1,1/m>; #end #end
#local test = ThreePointPlane(lp,mp,np);
#local isInside = inside(test,<0,0,0>);
#if (isInside = 0) object {test} #else object {test inverse} #end
#end
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
| |
|
|
"Samuel Benge" <stb### [at] THIShotmail com> schreef in bericht
news:46ccd5c4@news.povray.org...
> Hi everyone!
>
> No amazing eye candy to show you, but I have resolved the issues
> surrounding my inability to accurately depict odd angles for certain
> crystal morphologies.
>
> Attached is a png image showing both a screenshot from KrystalShaper,
> and a corresponding crystal shape rendered with POV-Ray. The problem
> areas are highlighted in red and green.
>
> I used exact angles and distances for this test. My reproduction seems
> 100% accurate, which means I can go on to create a library of minerals
> with support for randomized distances. A tutorial describing the
> technique may also be possible (although I still have that Weathered
> Surfaces tutorial to finish).
>
I love tutorials...... :-)
Thomas com> schreef in bericht
news:46ccd5c4@news.povray.org...
> Hi everyone!
>
> No amazing eye candy to show you, but I have resolved the issues
> surrounding my inability to accurately depict odd angles for certain
> crystal morphologies.
>
> Attached is a png image showing both a screenshot from KrystalShaper,
> and a corresponding crystal shape rendered with POV-Ray. The problem
> areas are highlighted in red and green.
>
> I used exact angles and distances for this test. My reproduction seems
> 100% accurate, which means I can go on to create a library of minerals
> with support for randomized distances. A tutorial describing the
> technique may also be possible (although I still have that Weathered
> Surfaces tutorial to finish).
>
I love tutorials...... :-)
Thomas
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Having thunk about it a little, I think one problem with using the Miller
indices directly is that the x,y,z axes are not always orthogonal in
crystal structures. For example, the x and y axes in the hexagonal system
perpendicular to them both. This goes for other structures too - e.g.
rhombohedral and monoclinic are bound to suffer similar problems. You might
have more luck with this approach for cubic systems.
Of course you could always define axis vectors for the different systems and
use them instead of the regular coordinate vectors.
On the other hand, you seem to be producing pretty nice crystal shapes with
your current method, so who am I to meddle with your productivity! :-)
Let's see some more renders like that first one
Bill
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Tim Attwood wrote:
> Cool, I saw your hexagon and was messing with
> Miller indices, and I found this site
> http://www.gly.uga.edu/schroeder/geol6550/millerindices.html
> which seems to explain simple Miller indices OK.
>
> Still haven't looked at Miller-Bravais indices.
>
> This is what I ended up with, but it's mostly untested.
I tried your macros, but they doesn't work for hexagonal structures.
They may work for cubic systems, but I still have yet to test 'em.
One would have to specify the so-called "unit cube" to work with other
crystal systems, in order to get the other axes right. Thanks for
contributing!
Sam
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Thomas de Groot wrote:
> I love tutorials...... :-)
I noticed :)
Now that I have no real work to do, I can concentrate on finishing up
that one...
Sam
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
| |
|
|
Bill Pragnell wrote:
> Having thunk about it a little, I think one problem with using the Miller
> indices directly is that the x,y,z axes are not always orthogonal in
> crystal structures. For example, the x and y axes in the hexagonal system
> perpendicular to them both. This goes for other structures too - e.g.
> rhombohedral and monoclinic are bound to suffer similar problems. You might
> have more luck with this approach for cubic systems.
I agree with everything you just said. All my (limited) research into
this area seems to point to the simple fact that:
> you could always define axis vectors for the different systems and
> use them instead of the regular coordinate vectors.
But I don't know how to implement this just yet. It *should* be as easy
yet to get it to work.
> On the other hand, you seem to be producing pretty nice crystal shapes with
> your current method, so who am I to meddle with your productivity! :-)
I think *any* crystal can be accurately depicted with this method, so
you could say I'm happy with it :) Punching in Miller indices would be
much easier, but hey, at least I can still squeeze geometry from that
demo version of KrystalShaper :)
> Let's see some more renders like that first one
I'll be making more, you can count on it :D
Sam
Post a reply to this message
|
|
| |
| |
|
|
|
|
| |
|
|